
Company X has gained 510(k) clearance for their new heart valve and was notified by the Food and Drug Administration (FDA) that post-market surveillance (PMS) is required. Why did they receive this notification, what does it mean, and how do they address it? You will know more after reading this blog.
Why?
There are several routes for manufacturers to conduct PMS. It may be ordered by the FDA under a “522 order.” PMS under a 522 order is covered in Title 21 Part 822 of the CFR, and is defined as an “active, systematic, scientifically valid collection, analysis, and interpretation of data or other information about a marketed device.” Section 522 of the Federal Food, Drug, and Cosmetic Act (FD&C Act) gives the FDA authority to require manufacturers to conduct PMS for certain class II and class III devices.
Section 522 of the Food Drug & Cosmetic Act states that:
“The Secretary may by order, at the time of approval or clearance of a device or at any time thereafter, require a manufacturer to conduct post-market surveillance for any device of the manufacturer that is a Class II or Class III device—
- the failure of the device would be reasonably likely to have serious adverse health consequences;
- that is expected to have significant use in pediatric populations; or
- that is intended to be—
- implanted in the human body for more than 1 year; or
- a life sustaining or life-supporting device used outside a device user facility.”
What happened before Company X received the PMS request? Before notifying a manufacturer, the FDA requested that a pre-522 team of experts review a situation and their review justified issuance of a 522 order. A reason for issuance could be a recent recall of a predicate device for unanticipated adverse events related to a predicate device. A 522 order includes information from the pre-market submission (e.g., innovation and what clinical data might be available), research questions to be answered, the rationale for the 522 order, and PMS plan design recommendations.
A total of 337 orders are registered on the FDA website and 25 orders are active to date. The number of orders per specialty per year are displayed in Figure 1. Interestingly, there are only a few years with a substantial number of orders issued. Is there a relation between the large number of orders issued in 2011 and 2012 and the health concerns on metal-on-metal hip implants and Poly-Implant Prothèse (PIP) breast implants?
The FDA National Evaluation System for Health (NEST) is a relatively new initiative to generate evidence across a product life cycle. Evaluation of real-world data (e.g., electronic patient records, national health registries) could help to identify safety issues more quickly.
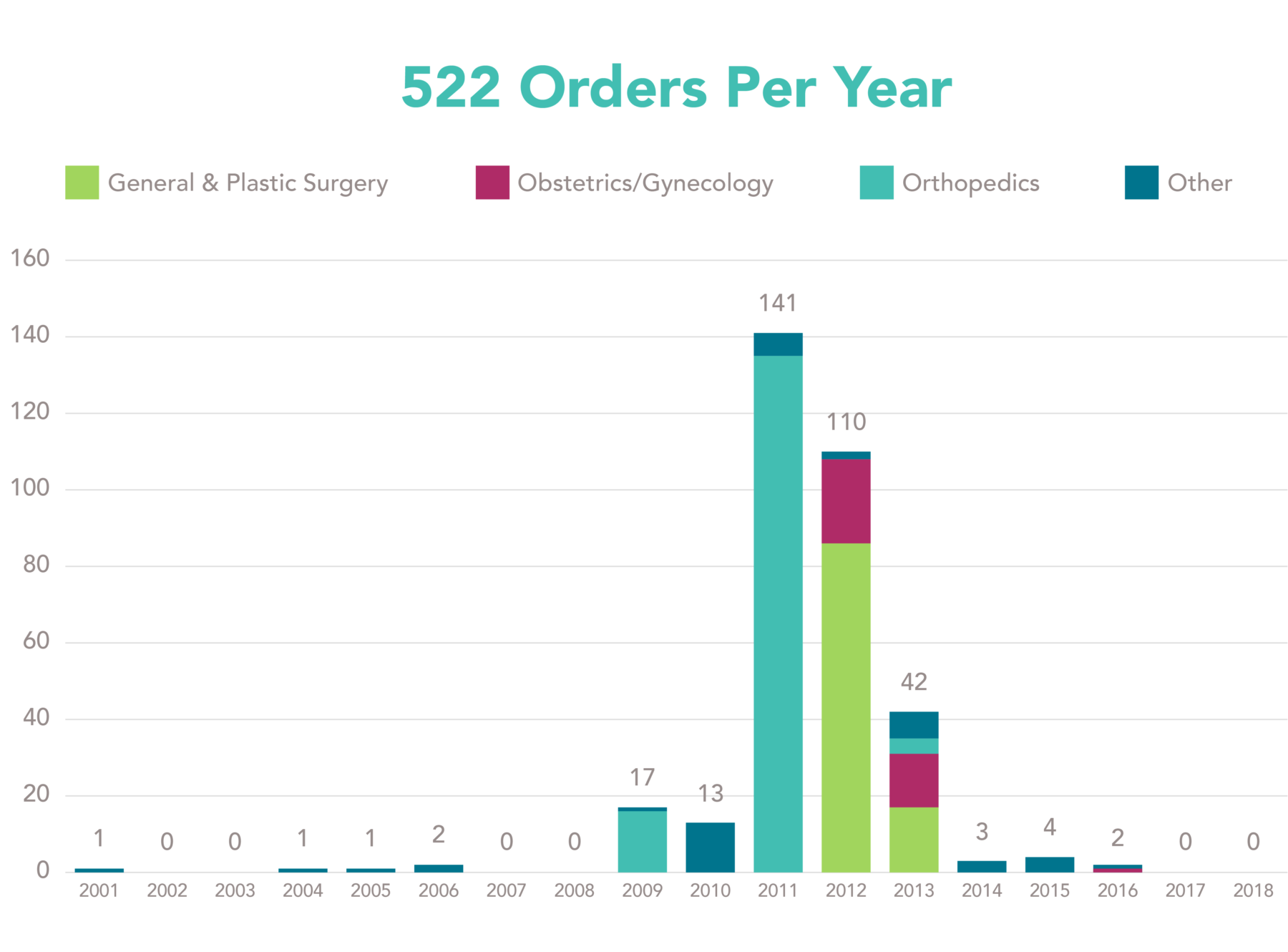
Figure 1: Number of 522 orders per specialty per year
What?
The PMS activities should be defined in a PMS plan and require approval by the FDA. The manufacturer should initiate PMS activities within 15 months after receipt of the 522 order. Interim and final PMS reports should be submitted to the FDA as approved and specified in the PMS plan. In case of unexpected findings (e.g., a high incidence of unanticipated adverse events), the FDA may request additional actions from the manufacturer. For instance, a high incidence of thromboembolic events related to a heart valve may require additional safety notification by the manufacturer to customers.
PMS of long-term implanted devices is considered important even after market suspension or market termination of a device, and a request by the manufacturer to discontinue PMS is rarely approved by the FDA. Further research may be required after completion of an order.
In Europe, PMS has become the focus of attention with the new Medical Device Regulation (MDR) that was adopted in 2017. Manufacturers are required to regularly evaluate and document clinical outcomes of their device to demonstrate continued safety and performance.
There are several PMS designs possible and the design that is ordered by the FDA will depend on quality and availability of existing clinical data. As such, lower level clinical study designs may be adequate to investigate a health concern.
How?
Procedures and requirements for PMS issued under Section 522 of the FD&C Act are implemented in Part 822 of Title 21 CFR. Guidance for PMS was issued in Post-market Surveillance Under Section 522 of the Federal Food, Drug, and Cosmetic Act, Guidance for Industry and FDA Staff with the latest version having been released on May 16, 2016. The document provides on overview of Section 522 of the FD&C Act, how to fulfill Section 522 obligations, and recommendations for design and review of a PMS plan.
A 522 order does not directly reflect the need to perform a clinical trial. It may also request additional preclinical testing, active or passive surveillance (e.g., data from the Manufacturer and User Facility Device Experience [MAUDE] or from national registries), or a scientific literature search on clinical outcomes of your device or a predicate device (meta-analysis and/or systematic review). A (new) clinical study may be required if existing clinical data is found inadequate to demonstrate safety of the device. In addition, data of previously performed pre- and post-market studies may be sufficient to answer research questions issued in the order. In other words, a well-maintained PMS system could help manufacturers to quickly respond to a 522-order, or even prevent one entirely.
Avania is an expert medical device CRO with broad experience in performing medical device clinical trials in the United States, Australia, and Europe. Our experts are available to define your PMS needs and help you with questions on post-market clinical trials. It’s time to follow up!